


杜卫民* 邓德华 韩志陶 肖伟 卞成 钱雪峰
安阳师范学院化学化工学院,安阳,河南,455002
摘要:以SnCl4·5H2O和CS2为前驱物,利用一种改进的溶剂热方法成功合成了六角形SnS2单晶纳米片,利用X射线衍射(XRD)、透射电子显微镜(TEM)和高分辨透射电子显微镜(HRTEM)对所得产物的物相、形貌以及晶格进行了表征。结果表明,所得SnS2纳米晶为具有六角片状形貌的单晶六方结构,其直径为20-70nm、厚度为7-10nm。结构和高分辨透射电镜分析表明,SnS2纳米片的六角片状形貌来源于其{001}晶面的生长受限以及其六个等能量、高指数的{110}晶面的快速生长。以罗丹明B(Rh B)的水溶液为模拟染料废水,进一步研究了所得SnS2六角纳米片在可见光条件下对染料的光催化降解性能。实验数据表明,所得SnS2纳米片对罗丹明B水溶液具有很好的光催化降解能力,在太阳光照射70分钟后的脱色率可达97.7%。良好的光催化性能表明,SnS2纳米片是一种性能优异的可见光催化剂,在环境保护、废水处理方面具有很好的应用前景。
Hexagonal tin disulfide nanoplatelets:A new photocatalyst driven by solar light
Du Weimin Deng Dehua Han Zhitao Xiao Wei Bian Cheng Qian Xuefeng
College of Chemistry and Chemical Engineering, Anyang Normal University, Anyang, Henan, 455002
ABATRACT:Single-crystalline, hexagonal tin disulfide (SnS2) nanoplatelets were successfully synthesized through an improved solvothermal process with SnCl4·5H2O and carbon disulfide (CS2) as precursors. The crystal phase, morphology, and crystal lattice of the prepared products were characterized by powder X-ray diffraction (XRD), transmission electron microscopy (TEM), and high-resolution transmission electron microscopy (HRTEM), respectively. Results reveal that the synthesized SnS2 nanoplatelets are in hexagonal structure with 20-60nm in diameter and 7-10nm in thickness. The structural and HRTEM analysis indicates that the formation of SnS2 nanoplatelets with hexagonal morphology results from the accelerating growth of six energetically equivalent high-index crystalline planes {110} and the retarded growth of {001} crystalline planes. The photocatalytic degradation properties of hexagonal SnS2 nanoplatelets driven by solar light were further investigated with Rhodamine B (RhB) as simulating pollutant. Results show that these SnS2 nanoplatelets have a good performance of photocatalytic degradation on RhB, and the decolorizing rate can reach 97.7% after irradiated for 70 minutes by solar light. Better photocatalytic properties indicate that hexagonal SnS2 nanoplatelets are a kind of promising photocatalyst driven by solar light and have potentially applied prospects in wastewater treatment and environmental protection.
1、引言
在现代社会的能源危机和环境污染问题面前,可见光催化剂有望通过利用太阳能分解水制氢和净化水与空气而有效解决这两个问题。尽管人们对二氧化钛已进行了很长时间的研究,但由于其能带隙较宽(锐钛矿:3.2ev;金红石:3.0ev),因此其只能吸收占太阳光很少部分的紫外光,限制了其在实际中的应用,因此,基于应用和经济的目的,需要去寻找能够有效利用太阳能的可见光催化剂。目前为止,已采用不同的策略用于二氧化钛的结构改性:如引进金属离子[1,2]、掺杂N、C、S等阴离子[3]以及与低能带的半导体材料复合[4]等。然而,这些掺杂的材料在可见光区域一般表现出的较小的吸收,导致其催化活性较低。所以,急需探索新型的光催化剂来有效解决二氧化钛在可见光下较低光催化活性问题。迄今为止,几种有希望的可见光催化剂已被尝试并取得了一些优异的结果。例如,InTaO4 [5]、BiFeO3 [6]、TaON [7]、Ag@AgBr [8]以及Bi2WO6 [9-11]等等。由此可见,新型光催化材料的探索是满足未来环境和能源技术要求的一种切实可行的途径。
作为一种n型半导体材料,SnS2属于层状金属硫属化物,具有六方的CdI2型的晶体结构。由于其层状结构中存在有大量的空位,SnS2成为一种重要的插层反应主体材料。实际上,该材料作为锂电池的插层电极已经被研究了很多年[12-14]。另外,由于合成的复合材料通常具有很多改进的性能(比如提高的电子、离子导电性),所以聚合物在SnS2中的插入成为目前研究的又一热点,已经有不同种类的聚合物被成功地插入到了SnS2材料中[15-17]。还有,SnS2具有较宽的能带隙(约为2.35eV)以及较强的各向异性的光学性质,因此,SnS2也可以被应用于其他的广泛领域,比如:气敏材料[18],光电设备[19],全记录系统及光学材料[20]等。
在过去的几十年里,由于其特殊的化学物理性能,各种从一维的到三维的各向异性的纳米材料吸引起了特别的关注[21, 22]。而且,由于其在磁学、光电学、杂相催化以及纳米电子学方面的应用,均匀的纳米材料自组装成一维到三维的超级结构也吸引了迅速增长的兴趣[23]。因此,纳米材料的形貌控制合成已受到了越来越多的注意。作为一种重要的半导体,人们对SnS2投入了较多的研究并获得了许多不同尺寸和形状的纳米材料。如,不规则的纳米粒子[24],花形的或芦荟形的纳米材料[25],纳米片[25, 27],纳米带[28, 29],薄膜[30, 31],纳米复合材料[32]等等。然而,与II?VI和III?V主族半导体纳米材料形貌控制合成的显著成果相比,所得的SnS2的形貌通常不规则且团聚现象严重。尽管也有几个优异的工作被报道,如:用氧化铝模板法合成的SnS2纳米管[33]、热分解单分子前驱物合成的纳米盘[14], 然而预制的氧化铝模板或有机金属前驱物的毒性限制了其在实际中应用。因此,如何利用一种简单廉价的方法制备具有可控形貌和较小尺寸的SnS2纳米材料仍然存在很大的困难。而且,相关的性能研究也可能为其在纳米设备方面的应用提供一些新的途径。
在目前的研究中,以SnCl4·5H2O和CS2为前驱物,利用一种改进的溶剂热方法成功合成了六角的SnS2单晶纳米片,据我们所知,这种以简单的无机盐作为原料合成的六角形SnS2单晶纳米片还鲜有报道。由于其的特殊结构和形貌,进一步研究了这种六角SnS2单晶纳米片的光学和可见光催化性能。
2、实验部分
2.1 六角形二硫化锡纳米片的合成
SnCl4·5H2O、CS2和苯甲醚均为分析纯试剂(购于阿拉丁化学试剂公司);油胺(80-90%)购于百灵威化学试剂公司。所有试剂均未做进一步处理而直接使用。在一个典型的合成过程中,将0.4mmol的SnCl4·5H2O和2mL的油胺在室温条件下加入到37mL的苯甲醚里面,将所得混合液加热到60~70oC并磁力搅拌约30分钟后,形成均一透明的溶液。将反应液倒入50mL的聚四氟乙烯容器中并加入1.0 mL的CS2,放入钢质的高压釜中密闭。于180℃反应24小时后,移出反应釜,自然冷却至50-60℃,离心分离,用无水乙醇反复洗涤,真空干燥即可得到SnS2纳米片。
2.2 结构表征
利用Rigaku D/Max-2200 X-射线衍射仪表征产物的物相:Cu Ka辐射源(λ=0.15418nm);样品的形貌及晶体结构通过透射电镜(JEOL JEM-2010, 加速电压为200kv)和高分辨透射电镜(HRTEM)(JEOL JEM-2100F, 加速电压200KV)进行观测。红外光谱在Perkin Elmer Paragon 1000傅里叶变换红外光谱仪上获得。
2.3光学和光催化剂性能测试
2.3.1 为了研究其光学性能,将SnS2纳米片超声分散于无水乙醇中,再进行紫外可见吸收光谱测量。其紫外可见吸收光谱是在室温下于Varian Cary 50紫外可见分光光度计上获得。
2.3.2 SnS2纳米片的光催化性能通过在太阳光下催化降解模拟污染物RhB水溶液进行评价。实验在室温下完成,具体过程如下:在一个典型的光催化剂实验中,将0.1g SnS2纳米片加入到100ml RhB溶液(10-5 mol·dm-3)中。在照射之前,将悬浮液在黑暗环境中磁力搅拌10分钟,以确保在光催化剂粉末和RhB之间建立吸附-脱附平衡。然后将溶液在磁力搅拌下暴露于日光照射中。在给定的时间间隔内,从反应溶液中取5 mL 悬浮液。将催化剂粉末和RhB溶液离心分离。借助F-722型紫外可见分光光度计检测其在553nm处的吸光度进行监控RhB浓度的变化。在整个实验过程中,太阳光的强度从62.5 W/m2变化到83.3W/ m2。为保证实验数据的可比性,所有平行实验均在相似的太阳光强度下平行完成。
3、结果与讨论
3.1 六角形二硫化锡纳米片的形貌与结构表征
透射电镜表明所得SnS2纳米材料呈现出典型的六边片状的纳米结构。图1a清晰地表明,当反应在180度反应24个小时后,SnS2纳米片是其主要产物。这意味着该方法可以大规模合成六角形SnS2纳米片。而且,当其重新分散在有机溶剂中并滴在铜网上时,这些SnS2纳米片在不同的区域会呈现两种取向:即平躺在底物上和面对面堆积形成柱状的纳米结构,这有利于对SnS2纳米片六角面和侧面的观察。电子衍射图谱中清晰的六方斑点表明所得SnS2纳米片为结晶良好的单晶结构。这些电镜照片表明所得SnS2纳米片的直径为20~60纳米,厚度为7~10纳米。图1c中实验样品的所有衍射峰完全符合标准六方的SnS2结构(JPCDS NO. 23-677),属P-3m1空间群,其晶胞参数为a = 3.649?和c = 5.899?,没有探测到其它杂质如SnS,Sn2S3,Sn3S4,Sn4S5、氧化物或有机物的峰,表明产物的纯净结晶。仔细观察实验所得XRD图谱可以发现,其(001)和(101)峰强度要比标准图谱里对应峰强度降低很多,而且还有一定程度的宽化现象。这意味着沿此晶面的生长受限以及特殊形貌的形成。
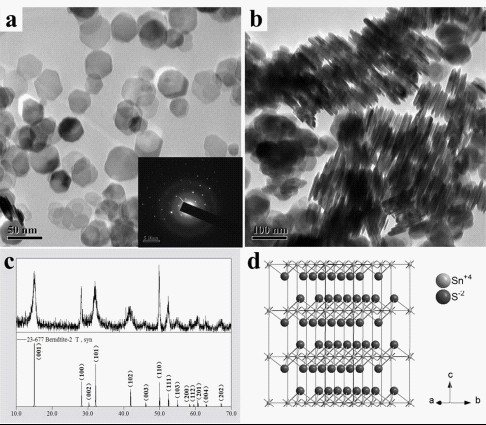
图1:(a, b)所得SnS2纳米片的不同放大倍数的透射电镜照片(图a中的插图为任意纳米片的电子衍射图谱);(c) SnS2纳米材料的X射线衍射图谱(上部分是实验所得图谱,下部分是卡号为23-677的标准图谱);(d) SnS2晶体的超晶格结构图:A = 3a,B = 3b, C = 3c。
材料晶体结构的图示化有助于进一步确定SnS2纳米片的形成控制因素。通过模拟体相SnS2晶体的XRD图谱,可以得到锡原子与硫原子的空间坐标(Sn1在0,0,01;S1在0.333,0.667,0.2600)[34]。基于SnS2材料的晶体参数以及各原子的空间坐标,可以模拟出SnS2的超晶格结构图(图1d:A=3a,B=3b,C=3c)。从图中可以发现,在SnS2晶体结构中,一层锡原子被上下两层硫原子包夹,形成一种CdI2型的“三明治”结构。而且,SnS2晶体沿C轴方向是由交替的SnS2 层和S-S 层交替组成。 据文献报道,在SnS2 层内的作用为共价键,而在S-S 层之间的作用为较弱的范德华力[35]。因此其C轴方向的生长极易受到外在因素的影响。考虑到SnS2的空间结构,可以认为SnS2内在的各向异性结构特性控制了初级SnS2粒子(即片状晶种)的形貌,进而影响了SnS2沿a,b,c轴方向的生长速率。同时,所得SnS2纳米片的红外图谱(图2)表明,油胺在所得纳米材料的表面有明显的吸附作用。因此,这种油胺在SnS2纳米晶表面的修饰进一步限制了其 <001> 方向的生长,扩大了纳米片上下表面和侧面之间的能量差,导致了SnS2六角片状形貌的形成。
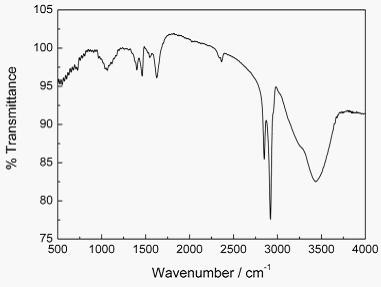
图2:SnS2纳米片典型样品的红外光谱图
3.2 高分辨电镜分析
为了进一步认识SnS2的片状晶体结构,我们对两种纳米片进行了高分辨透射电镜分析:一种是平铺的纳米片,另一种是竖立形成堆积纳米柱状体的纳米片。不同朝向纳米片的清晰晶格图像表明所得SnS2纳米片为单晶结构。一个平铺的六角纳米片的高分辨透射电镜照片清楚地表明,其1.80?和3.14?的晶格间距分别对应于六方SnS2的(110)和(100)面的晶面间距值(图3b)。从图3d可以发现,5.87?的晶格间距与SnS2(001)面的晶面间距值相同,说明六角SnS2纳米片优先沿其六角面进行堆积,即<001>方向,而不是其侧面。以上结果表明SnS2纳米片为单晶结构,其<001>方向是生长最慢的方向。因此, SnS2纳米片六角片状形貌的形成来源于其{001}晶面的生长受限以及其六个等能量、高指数的{110}晶面的快速生长。这与我们先前报道的CuS纳米片的形成机理非常相似[36]。

图3:(a)平铺的SnS2六角纳米片的透射电镜照片;(b)图a中白框所围区域的高分辨透射电镜照片,电子束由<001>方向入射;(c)几个竖立的六角纳米片的透射电镜照片;(d)图c局部的高分辨透射电镜照片,电子束由<010>方向入射
3.3光学性能
一般而言,由于其较小的尺寸和增加的能带隙,半导体纳米晶的紫外可见吸收光谱与其体相材料相比会表现出明显的“蓝移”现象。但是其物理性质同样受到表面形态的强烈影响。特别是,当半导体纳米颗粒的表面被另一种物质化学修饰后,如果它们之间的介电常数差异足够大,将会表现出介电限域效应。因此,当由表面形态引起的能量变化大于由尺寸效应引起的能量变化时,纳米晶的能带隙将会减少,材料的紫外可见光吸收光谱将会显示出“红移”而不是“蓝移”[37]。
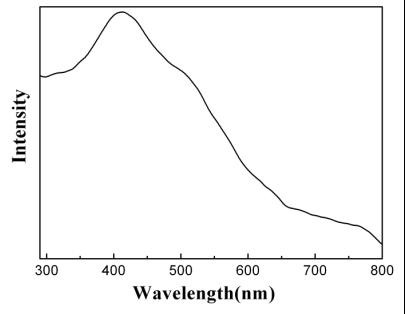
图4:SnS2 纳米片的室温紫外可见吸收光谱
图4是180oC合成的SnS2纳米片的紫外可见光吸收光谱。从图中SnS2纳米片的吸收边缘估计材料的能带隙为1.91eV。文献报道的体相二硫化锡的能带隙为2.35eV,对应的紫外可见吸收为~528 nm。考虑到SnS2纳米片在紫外可见吸收光谱的起始吸收位置(~650nm),其吸收位置的“红移”现象意味着在获得的产物中存在着激子跃迁的介电限域效应。由于在目前的工作中油胺被用作包覆剂,所以获得的SnS2纳米片外面包有一层有机表面活性剂。而且,油胺是一种极性很小的有机物,SnS2是一种具有较大介电常数的高电荷物质。因此,这种介电常数的较大差异导致了SnS2纳米片紫外可见吸收光谱上的“红移”现象。这也导致二硫化锡纳米片在300—650nm间较宽的吸收,有利于其在太阳光下的光催化性能。
3.4 光催化性能
由于其特殊的结构与形貌,所得SnS2纳米片被选为一种潜在的可见光催化剂。在先前的报道中,可见光催化剂通常需要以氙灯作为模拟太阳光进行激发。虽然也可以得到一些较好的数据,但是在真实太阳光下的光催化结果将更具有实现的应用意义。因此,所得SnS2纳米片的光催化性能通过在太阳光下光催化降解Rh B的水溶液进行评价。
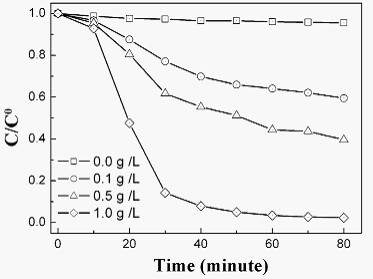
图5:罗丹明B(Rh B)溶液在不同催化剂含量实验条件下的相对浓度随照射时间的降解曲线
图5为罗丹明B溶液在不同SnS2纳米片含量条件下的相对浓度随照射时间的降解曲线,其中C/Co是Rh B的相对浓度。根据朗伯比尔光学定律,稀染料溶液的浓度与其吸收强度成比例。所以以下等式可以被推出:C/Co = A/Ao。C & A是染料在不同时间的浓度和其最大吸收波长处的吸收强度;Co & Ao是溶液的起始浓度和吸收强度。空白试验表明Rh B在太阳光下的自身降解可以忽略不计。十分钟的实验数据是在暗处通过超声使SnS2纳米片和染料达到吸附-脱附平衡时的Rh B的相对浓度。其相对浓度的稍微降低来源于染料在光催化剂表面的吸附。由图可以看出,其可见光催化效果随着SnS2纳米片含量的增加而提高。在SnS2纳米片含量为0.1 g/L、0.5 g/L和1.0 g/L时,其最大的脱色率(1- C/Co)分别为40.5%、60.4%和97.7%。光照七十分钟后,SnS2纳米片含量为1.0g/L的罗丹明B的相对浓度减少到几乎为零。这个结果和溶液从粉红色变为无色的实验现象非常吻合。这些结果表明,获得的SnS2纳米片对罗丹明B有很好的光催化活性。
图6是不同实验条件下Rh B的对比试验结果。由图可见即使将Rh B的浓度加倍,SnS2纳米片仍然保持了较高的催化活性,其最大脱色率达91.9%。这也表明1.0 g/L的SnS2纳米片可以充分吸收太阳光,产生优异的光催化性能。作为对比,还按照传统的方法合成了体相的SnS2材料。当1.0 g/L的体相SnS2材料作为光催化剂时,在同样的太阳光强度和辐照时间时,其最大脱色率只有66.8%。
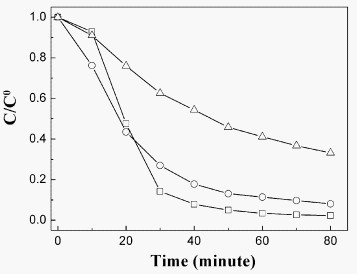
图6:不同实验条件下罗丹明B 的相对浓度变化与照射时间的关系: (“) C0BhB = 1×10-5mol·L-1, 1.0 g/L SnS2 纳米片为催化剂 (O) C0BhB = 2×10-5mol·L-1, 1.0 g/L SnS2纳米片为催化剂. (Δ) C0BhB = 1×10-5mol·L-1, 1.0 g/L体相SnS2为催化剂.
根据传统的光催化理论,染料的氧化反应一般认为是靠羟基自由基和氧自由基的引发[4]。因此,避免电子空穴对的复合对于持续产生羟基自由基和氧自由基非常重要。紫外可见光谱上较宽的吸收使SnS2纳米片对太阳光具有较好的吸收。而且由于其尺寸较小,光生的电子空穴对将很快扩散到材料的表面,使材料内部的电子空穴复合几率大大减少。结果,较多的光生电子和空穴可被用于光催化反应中,使模拟污染物迅速降解。另外,由于其片状的纳米结构以及纳米材料的表面效应,SnS2纳米片较大的比表面积也有利于其光催化性能。较大的比表面积不仅为有机化合物的降解反应提供更多的活性位点,还可以有效促进电子空穴对的分离效果。当较低含量的SnS2纳米片作为催化剂时,吸附的染料分子数量降低,所以光子不能被有效地利用,其脱色率较低。随着光催化剂含量的增加,催化活性中心倍增,脱色率上升。当SnS2纳米片的量增加到一定程度,其吸收能力达到饱和,催化活性就会被维持恒定。如果加入过量的催化剂,将会遮蔽太阳光,降低催化反应效果,浪费催化剂。一句话,二硫化锡纳米片优异的光催化效果来源于其较宽的可见光吸收、较小的尺寸、较大的比表面积。
4、结论
总之,以简单无机盐为原料,通过一种简单的方法成功地合成了大量的六角形SnS2纳米片。值得注意的是所得六角形SnS2纳米片表现出优异的光催化性能,在太阳光下可以很好的催化降解罗丹明B。六角形SnS2纳米片特殊的结构和形貌决定了其优异的光催化性能:即较宽的可见光吸收、较小的尺寸、较大的比表面积。这种六角SnS2纳米片将来很可能在降解染料废水方面成为一种高效的可见光催化剂。
参考文献
[1]. H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, K. Ikeue and M. Anpo, J. Photochem. Photobiol. A, 2002, 148, 257-261.
[2]. M. Kitano, M. Takeuchi, M. Matsuoka, J. Thomas and M. Anpo, Catal. Today, 2007, 120, 133-138.
[3]. R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269-271.
[4]. G. Li, D. Zhang and J. C. Yu, Environ. Sci. Technol., 2009, 43, 7079-7085.
[5]. Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2002, 414, 625-627.
[6]. F. Gao, X. Y. Chen, K. B. Yin, S. Dong, Z. F. Ren, F. Yuan, T. Yu, Z. G. Zou and J. M. Liu, Adv. Mater., 2007, 19, 2889-2892.
[7]. G. Hitoki, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Commun., 2002, 2, 1698-1699.
[8]. P. Wang, B. Huang, X. Zhang, X. Qin, H. Jin, Y. Dai, Z. Wang, J. Wei, J. Zhan, S. Wang, J. Wang and M.-H. Whangbo, Chem. Eur. J., 2009, 15, 1821-1824.
[9]. H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432-22439.
[10]. Z. Chen, L. Qian, J. Zhu, Y. Yuan and X. Qian, CrystEngComm, 2010, 12, 2100-2106.
[11]. M. Shang, W. Wang, S. Sun, L. Zhou and L. Zhang, J. Phys. Chem. C, 2008, 112, 10407-10411.
[12]. T. Momma, N. Shiraishi, A. Yoshizawa, T. Osaka, A. Gedanken, J. Zhu and L. Sominski, J. Power Sources, 2001, 97-98, 198-200.
[13]. X.-L. Gou, J. Chen and P.-W. Shen, Mater. Chem. Phys., 2005, 93, 557-566.
[14]. J. W. Seo, J. T. Jang, S. W. Park, C. J. Kim, B. W. Park and J. W. Cheon, Adv. Mater., 2008, 20, 4269-4273.
[15]. E. Aharon, A. Albo, M. Kalina and G. L. Frey, Adv. Funct. Mater., 2006, 16, 980-986.
[16]. P. J. Ollivier, T. E. Mallouk, N. I. Kovtyukhova and S. W. Keller, Chem. Commun., 1998, 1563-1564.
[17]. H. Morales, H. Santos, H. R. R. Barrado and H. P. Espino, J. Solid State Chem., 2000, 398, 391-398.
[18]. W. D. Shi, L. H. Huo, H. S. Wang, H. J. Zhang, J. H. Yang and P. H. Wei, Nanotechnology, 2006, 17, 2918-2924.
[19]. N. Takeda and B. A. Parkinson, J. Am. Chem. Soc., 2003, 125, 5559-5571.
[20]. Y. Bertrand, A. Barski and R. Pinchaux, Phys. Rev. B, 1985, 31, 5494.
[21]. C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025-1102.
[22]. Y. N. Xia, P. D. Yang, Y. G. Sun, Y. Y. Wu, B. Mayers, B. Gates, Y. D. Yin, F. Kim and Y. Q. Yan, Adv. Mater., 2003, 15, 353-389.
[23]. Y. Huang, X. F. Duan, Q. Q. Wei and C. M. Lieber, Science, 2001, 291, 630-633.
[24]. B. Hai, K. B. Tang, C. R. Wang, C. H. An, Q. Yang, G. Z. Shen and Y. T. Qian, J. Cryst. Growth, 2001, 225, 92-95.
[25]. H. L. Zhu, X. Ji and D. R. Yang, J. Mater. Sci., 2006, 41, 3489-3492.
[26]. G. Shen, D. Chen, K. Tang, L. Huang, Y. Qian and G. Zhou, Inorg. Chem. Commun., 2003, 6, 178-180.
[27]. D. Chen, G. Shen, K. Tang, S. Lei, H. Zheng and Y. Qian, J. Cryst. Growth, 2004, 260, 469-474.
[28]. Y. J. Ji, H. Zhang, X. Y. Ma, J. Xu and D. Yang, J. Phys.: Condens. Matter, 2003, 15, L661-L665.
[29]. D. K. Ma, W. Zhang, Q. Tang, R. Zhang, W. C. Yu and Y. T. Qian, J. Nanosci. Nanotechnol., 2005, 5, 806-809.
[30]. G. Barone, T. G. Hibbert, M. F. Mahon, K. C. Molloy, L. S. Price, I. P. Parkin, A. M. E. Hardy and M. N. Field, J. Mater. Chem., 2001, 464 - 468.
[31]. T. G. Hibbert, M. F. Mahon, K. C. Molloy, L. S. Price and I. P. Parkin, J. Mater. Chem., 2001, 469 - 473.
[32]. G. Barone, T. Chaplin, T. G. Hibbert, A. T. Kana, M. F. Mahon, K. C. Molloy, I. D. Worsley, I. P. Parkin and L. S. Price, J. Chem. Soc., Dalton Trans., 2002, 1085 - 1092.
[33]. D. Chen, G. Z. Shen, K. B. Tang, Y. K. Liu and Y. T. Qian, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 747-749.
[34]. F. A. S. Al-Alamy, A. A. Balchin and M. White, J. Mater. Sci., 1977, 12, 2037-2042.
[35]. H. J. Chang, E. J. In, K. J. Kong, J. O. Lee, Y. M. Choi and B. H. Ryu, J. Phys. Chem. B, 2005, 109, 30-32.
[36]. W. Du, X. Qian, X. Ma, Q. Gong, H. Cao and J. Yin, Chem. Eur. J., 2007, 13, 3241-3247.

